zespół marfana
Jesteś wysoki? A może znasz kogoś takiego ? Zazwyczaj szczupły, wiotki, ma giętkie stawy (człowiek guma), może ma też piękne dłonie pianisty (o długich palcach)? A szczupła twarz i opadające kąciki oczu, do tego często okulary? Problemy ortopedyczne – skolioza i inne ? To też brzmi znajomo?
Ktoś kto ma takie cechy może nawet nie wiedzieć, że ma chorobę tkanki łącznej. Tkanka łączna to także naczynia tętnicze. To co najniebezpieczniejsze w chorobach tkanki łącznej to BRAK WIEDZY! Poszerzające się tętnice nie dają żadnych objawów! TĘTNIAK ROŚNIE W CISZY!
Aktualności Stowarzyszenia Marfan Polska




Główne cechy zespołu Marfana i innych marfanopodobnych chorób tkanki łącznej
Poniżej prezentujemy główne cechy zespołu Marfana i tym podobnych chorób tkanki łącznej. Jeżeli rozpoznajesz u siebie kilka z nich – niezwłocznie zgłoś się do lekarza, zawsze pilnie do kardiologa oraz planowo do lekarza genetyka. Doświadczony lekarz genetyk dzięki badaniu przedmiotowemu oceni czy może to być choroba tkanki łącznej.
Zauważ, że niektóre z tych cech są widoczne gołym okiem, a innych w ogóle nie widać. Dotyczy to najgroźniejszego – tętniaka. Dlatego Stowarzyszenie Marfan Polska wszystkim pacjentom zawsze zaleca pilną konsultację u lekarza kardiologa i co najważniejsze – wykonanie echo serca, bo #TętniakRośnieWciszy.

Wysoka, szczupła sylwetka

Elastyczne stawy

Długie ramiona, nogi i palce

Wady wzroku

Tętniak i rozwarstwienie aorty

Wady zastawek serca

Skrzywienie kręgosłupa - skolioza

Wady klatki piersiowej





Odma i inne choroby płuc

Przewlekłe bóle

Nadmierna męczliwość

Zwyrodnienie stawów
od czego zacząć?
1. Wizyta u lekarza POZ
Zawsze trzeba zacząć od wizyty u lekarza POZ (Podstawowa Opieka Zdrowotna), celem wydania skierowania do lekarza specjalisty: kardiologa (pilne) i genetyka (planowe). Ważne, aby lekarz rodzinny miał kontrakt z NFZ. W przeciwnym razie wydane przez niego skierowanie nie będzie ważne. Poinformuj lekarza o swoich podejrzeniach dot. zespołu Marfana. Jeżeli lekarz odmawia pomocy skontaktuj się z nami. Możesz też poprosić o pisemną odmowę wydania skierowania wraz z uzasadnieniem.
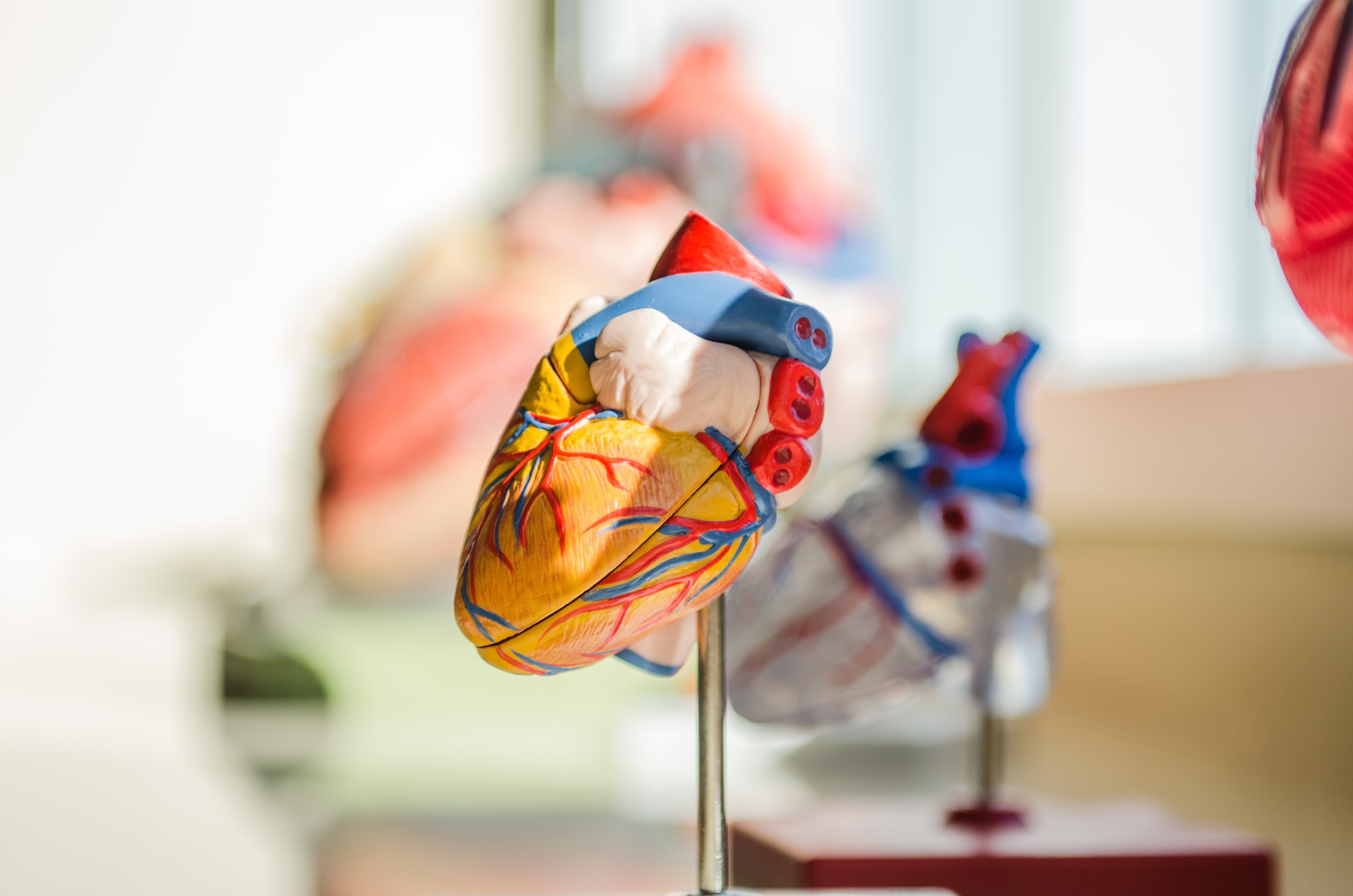
Pilna konsultacja z kardiologiem i wykonanie echo serca
Kardiolog oprócz przeprowadzania wywiadu, osłuchania pacjenta i zmierzenia ciśnienia, powinien pilnie wykonać lub zlecić wykonanie badania echo serca. Pamiętaj, że badanie echo serca inaczej USG serca to nie to samo co EKG – nie będzie na nim widocznych poszerzeń aorty ani niedomykalności zastawek.
Jak powinno wyglądać badanie echo serca
Badanie echo jest całkowicie bezpieczne i bezbolesne. Wygląda bardzo podobnie jak USG, tyle, że badanym organem jest serce. Podczas badania bardzo ważne jest, by lekarz wykonał pomiary wszystkich struktur serca, zarówno zastawek jak i aorty zwłaszcza opuszki i aorty wstępującej. Wyniki pomiarów powinny być zapisane w milimetrach oraz w skali z-score co ważne szczególnie u dzieci, bo skala z-score określa nie tylko sam wymiar ale jego odniesienie do wieku, wagi i wzrostu dziecka. U zdrowego pacjenta wskaźnik ten powinien wynosić ok. 1, jeżeli wskaźnik ten jest wyższy niż 2 to konieczna jest dalsza diagnoza i kontrola. Ważne – wynik badania echo serca musisz dostać na własność.
Wskaźnik Z-Score można sprawdzić na stronie www.marfan.org/dx/zscore
Genetyk
Konsultacja u lekarza genetyka powinna odbyć się w ośrodku i u lekarza, który ma doświadczenie w badaniu pacjentów z chorobami tkanki łącznej. Zespół Marfana i inne zespoły marfanopodobne to choroby rzadkie, a więc rzadko spotykane. Większa jest szansa na trafną diagnozę jeśli lekarz widział i badał większą ilość pacjentów z podobnym schorzeniem. Badanie takie powinno zawierać szczegółowy wywiad rodzinny, choroby i zdarzenia kardiologiczne u przodków, cechy ich budowy, okoliczności śmierci jeśli już nie żyją.
Drugim elementem jest badanie przedmiotowe czyli oglądanie pacjenta. Lekarz genetyk powinien wykonać m. in.ocenę stanu skóry, mięśni, stawów, zwrócić uwagę na ich zwiększoną ruchomość, wykonać test kciuka i test nadgarstka. Powinien także dokonać dokładnych pomiarów wzrostu i wagi pacjenta, rozpiętości ramion. Jeśli stosunek rozpiętości ramion jest większy niż wysokość ciała pacjenta to może wskazywać na zespół Marfana. Lekarz oceni też wady ortopedyczne: skoliozę, asymetrie lub wady klatki, wady stóp i kolan. W ocenie stanu pacjenta lekarz weźmie także pod uwagę przebieg rozwoju, zwłaszcza jeśli badane jest dziecko. Jeśli przed wizytą u genetyka wykonane było badanie echo serca, badania u okulisty, RTG kręgosłupa, klatki piersiowej lub płuc – koniecznie zabierz wyniki tych badań na wizytę. Będą one ważnym elementem diagnostycznym. Na koniec wizyty lekarz genetyk powienien ocenić prawdopodobieństwo wystąpienia zespołu Marfana lub innej choroby tkanki łącznej.
Poproś lekarza o wystawienie karty informacji genetycznej, będzie to istotne dla Twojego dalszego funkcjonowania z chorobą. Istnieje taka możliwość, że lekarz genetyk na podstawie objawów nie będzie w stanie potwierdzić lub wykluczyć choroby. Możliwe, że skieruje Cię wówczas na badania genetyczne czyli testy mutacji wykonywane z próbki krwi. Nie wszystkie poradnie genetyczne mają możliwość przeprowadzania takich testów. Nie są one refundowane w ramach koszyka świadczeń, ale na szczęście w coraz większej ilości ośrodków, zwłaszcza klinicznych są programy badań naukowych, dzięki którym pacjenci mają za darmo przeprowadzone takie badania.
Badania genetyczne
Diagnozę zespołu Marfana, tak jak innych zaburzeń tkanki łącznej podejrzewać może lekarz klinicysta, jednak potwierdzić ją można jedynie analizą materiału genetycznego. W chwili obecnej jest wiele metod umożliwiających diagnostykę genetyczną. Najbardziej powszechnie stosowane, jest klasyczne sekwencjonowanie metodą Sangera, jednak obecnie coraz bardziej popularne są panele NGS oraz WES i WGS.
Dobór odpowiedniego badania genetycznego powinien być przedyskutowany zarówno przez lekarza kierującego, jak i przemyślany przez pacjenta. W przypadku wielu cech klinicznych mogących świadczyć o Zespole Marfana, można rozpocząć identyfikację, od analizy genu FBN1 metodą Sangera. Jeżeli wynik badania molekularnego jest odmienny od oczekiwań lekarza, może on zlecić sekwencjonowanie określonych paneli DNA metodami II generacji (NGS).
Chorób tkanki łącznej jest wiele, część objawów wspólna, a jednoznaczna ich klasyfikacja możliwa jest dzięki sekwencjonowaniu wielu znanych genów je warunkujących. Pomocne są także metody sekwencjonowania WES i WGS, dzięki którym możliwa jest wielkoskalowa analiza całych genów. WES i WGS to metody niezastąpione u pacjentów, u których występuje wiele, zróżnicowanych objawów, na podstawie których trudno jest wyznaczyć lekarzom konkretną ścieżkę diagnostyczną. Badania WES i WGS to najszybszy sposób na znalezienie przyczyny nietypowej choroby, co znacznie przyspiesza przejście przez tzw. odyseję diagnostyczną. Metody te stają się również stosunkowo coraz tańsze niż w porównaniu do tradycyjnych badań Sangerowskich.
czytaj więcej o badaniach genetycznych
Gdzie można wykonać badania genetyczne
Pracownia Cytogenetyczna – Katedra i Zakład Genetyki Klinicznej AM
Prof.dr hab. n. med. Olga Haus
ul. M. Skłodowskiej-Curie 9
85-094 Bydgoszcz
Pracownia Genetyki
52 585 35 68
52 585 35 67
52 585 35 77
BIAŁYSTOK
Zakład Genetyki Klinicznej Akademii Medycznej
ul. Waszyngtona 13
15-089 Białystok
85 748 59 80
85 748 59 92
genetyka@umb.edu.pl
GDAŃSK
Katedra – Zakład Biologii i Genetyki Akademii Medycznej w Gdańsku
Prof. dr hab.n.med. Janusz Limon jlimon@amg.gda.pl
ul. Dębinki 1
80-211 Gdańsk
58 349-15-31
Katgen@amed.amg.gda.pl
Poradnia Genetyczna Konsultant dr Jolanta Wierzba
ul. Kliniczna 1a
80-402 Gdańsk
58 349 34 68
KATOWICE
Katedra i Zakład Biologii Ogólnej, Molekularnej i Genetyki Śl. AM Kierownik Prof. dr hab. n. med. Aleksander Sieroń
ul. Medyków 18
40-752 Katowice,
32 208 83 94
alsieron@sum.edu.pl
ŁÓDŻ
Zakład Genetyki Instytutu CZMP w Łodzi Kierownik dr hab. med. Lucjusz Jakubowski
ul. Rzgowska 281/289
93-338 Łódź
42 271-11-83
SZCZECIN
Zakład Genetyki i Patomorfologii PAM
Prof. dr hab. med. Jan Lubiński lubinski@rl.pam.szczecin.pl
Al. Powstańców Wkp. 72
70-111 Szczecin
91 482-24-21 w. 112, 113
WARSZAWA – CZD
Zakład Genetyki Medycznej Instytutu „Pomnik – Centrum Zdrowia Dziecka”
Prof. dr hab. med. Krystyna Chrzanowska chrzanowska@czd.waw.pl
Al. Dzieci Polskich 20
04-736 Warszawa-Międzylesie
22 815 74 52
genetyka@czd.pl
WARSZAWA – MEDGEN
WARSZAWA – IKARD
Ośrodek Badań Przesiewowych Dziedzicznych Chorób Układu Sercowo-Naczyniowego. Instytut Kardiologii w Aninie.
WROCŁAW
Katedra Patofizjologii – Zakład Genetyki
Dr hab. med. Maria Sąsiadek
ul. Marcinkowskiego 1
50-368 Wrocław
71 320-99-91, w. 980; 320-99-89
genetyka@gen.am.wroc.pl
Konsultacje dodatkowe
Warto wykonać dodatkowe konsultacje, zwłaszcza jeżeli obserwujesz u siebie jakiekolwiek objawy z danego układu. Zrób to zanim pójdziesz na wizytę do lekarza genetyka, wówczas w poradni genetycznej będzie większa szansa na potwierdzenie lub wykluczenie podejrzenia choroby.
Stowarzyszenie Marfan Polska zaleca konsultacje u następujących specjaistów:
- rehabilitant
- okulista
- ortopeda
- pulmonolog jeśli masz epizody astmy, duszności lub duże deformacje klatki piersiowej
- torakochirurg jeśli są wady klatki piersiowej
- endokrynolog bo u dzieci możliwa jest terapia hamowania wzrostu, ale tylko we wczesnym wieku
WSPIERAJĄ NAS:





")